


Voyager: Al Sandrock
“All it takes is to meet one patient who has benefited from a drug you helped to develop.”

Medicine, like sailing, can be an unpredictable vocation. Careful planning, and a skillful crew, can greatly increase the chance of success. Yet in both professions, circumstances change at a moment’s notice. The physician William Osler speaks to this nautical connection: “To study the phenomena of disease without books is to sail an uncharted sea, while to study books without patients is not to go to sea at all.”
Al Sandrock MD PhD, CEO of Voyager Therapeutics and son of a merchant marine captain, adds a more concrete link: “when you are at sea, people also come to the captain with their medical problems. When [my father] was able to help someone with their problem, he felt so great.”
With his father’s impromptu medical experiences buoying his interests, Sandrock decided to become a physician. Of all the disciplines, neurology and psychiatry most resemble the “uncharted sea.” Mapmaking is laborious—with billions of neurons to explore, recent work has shown that just a cubic millimeter of human brain contains 150 million synapses or connections (1.4 petabytes of data).
As an undergraduate at Stanford, Sandrock was exposed to the expansive mystery of neuroscience and disease: “I remember a lecture by the psychiatrist Jack Barchus…where he discussed the biological basis of schizophrenia and depression. I was blown away that you could explain such complex diseases by imbalances in chemicals in the brain.”
Determined to create new therapies for patients suffering from neurologic disease, Sandrock pursued MD-PhD training from Harvard medical school. With scientific pillars like Paul Patterson, Story Landis, Ed Furshpan, and David Potter for support, he studied axon guidance and regeneration in the context of nerve injury: “Why can peripheral nerves regenerate [to an extent] whereas central neurons do not? Even my PhD was geared towards addressing clinical problems. I had this early desire to help advance new therapies,” he describes.
As a resident neurologist at Mass General Hospital [MGH], Sandrock witnessed how better “compasses” helped neurologists navigate disease: “Even in the 1990s we started to have some tools at our disposal.” Specifically, he witnessed how gadolinium enhanced MRIs could help distinguish new vs residual lesions in multiple sclerosis (MS). Learning from experts like Steve Hauser and (his co-resident) Tim Vartanian, Sandrock saw how MRI lesions could be used as a surrogate marker for disease activity—crucial to evaluating new therapies like beta-interferon: “The effect of beta-interferon on reducing new or enlarging lesions was really what made many of us really believe in its effect. Here was objective surrogate evidence that correlated with the clinical results.”
Seeing the betaseron story unfold inspired the young neurologist to learn professional drug development. Leaning on his former co-resident Nancy Simonian for support, Sandrock left his prestigious NIH grants and Harvard tenure track job to join Biogen. The decision was an agonizing one: “I initially declined the offer and kept my grants active…but Nancy really went to bat for me, and I was able to get the job [offer] back. I started in February of 1998.”
What followed was arguably one of the most prolific drug development careers in biotech—certainly within the field of neuroscience. Under Sandrock’s captainship, the crew at Biogen developed drugs like Tysabri, Tecfidera, Plegridy, Spinraza and Aduhelm. Despite the successes, Sandrock speaks first to his shortcomings: “I have failed at certain points in my career and been at times even been dragged through the mud for it…failure is the nature of our industry.” He goes on to say: “all it takes to keep going is to remember the patients you serve…I have met children who would not be alive today without Spinraza. I know MS patients who [with the help of our treatments] could become moms and enjoy their children…I still feel like a neurologist, but my ‘practice’ now serves a larger population.”
This desire to serve a larger population, and again captain the ship, led Sandrock to the CEO position at Voyager Therapeutics in 2022. Put simply, Voyager seeks to build better boats for neurology drug development. The hull of the company is Voyager’s brain penetrant AAV capsid technology. One family of Voyager’s novel capsids utilizes the ALPL receptor to cross the BBB into the CNS—this technology is designed to allow gene therapies to access difficult to reach areas of the CNS (e.g. deep brain structures) without the steep concentration gradients of intrathecally injected oligonucleotide treatments. In our interview, Sandrock describes how the Voyager scientists discovered the capsids (TRACER platform), identified ALPL and other receptors, and the suite of medicines and modalities they are pursuing. Their pipeline includes monoclonal antibodies (Tau/AD), AAV-delivered vectorized siRNA (SOD1/ALS, Tau/AD), vectorized antibodies (Aβ/AD), and gene-replacement therapies (FXN and GBA1 in partnership with Neurocrine).
In our discussion, Sandrock highlights his guiding principles for navigating clinical development (the Sandrock Shamrock Q#6) and underscores the obstacles that can sink neurology drugs before they get to patients. Eager to give credit to his crew, Sandrock stresses the team-based nature of drug development: “It takes so much expertise to build a drug that teamwork is a necessity—it cannot be “all about you,” and credits Voyager scientists with persisting in identifying the ALPL receptor (Q#8).
Voyager is pushing the boundaries of neurogenetic medicines. They are sailing into the wind: it is a high-risk, high-reward journey. With Sandrock at the helm, and recent hires expert in neuro drug development (Toby Ferguson/CMO and Nate Jorgenson/CFO), they stand a good chance of calming the choppy waters. My hope is that they can (finally) ferry neurology patients in the right direction.
Below is an interview with Al Sandrock, CEO of Voyager Therapeutics from August 2024:

1. What initially made you interested in science and medicine, and led you to the HMS MD-PhD program? Any early mentors you want to highlight?
My parents were a big influence on my decision to pursue medicine. My father joined the merchant marines when he was 16 years old, so he did not finish high school. My mother grew up in Japan, and in many ways was trained to be a wife and mother. But my parents both revered physicians…just revered them. My father eventually became captain of a ship and would always tell me: “when you are out at sea, people come to the captain with their medical issues.” He would pull out the Merck Manual and do his best. When he was able to help someone with their problem, he told me that he felt so great. This had a big impression on me as a child.
In high school, I remember seeing a film where a scientist dissected a frog leg and made a muscle prep. The idea that electricity could stimulate muscle contraction was amazing. I found it unbelievable that science could help understand this process. This cemented a dual love of both biology and medicine. When I was in college, my father’s brother died in his 40s from heart disease. I remember realizing that there is so much left to be done in terms of making new medicines. Even when I was just starting medical school, I had a desire to help make new drugs.
In college [at Stanford], Donald Kennedy was the head of the human biology department. He was one of the pillars of neuroscience in the United States in the early days, who ended up becoming President of Stanford. He gave lectures on the nervous system that were just beautiful. He would invite physicians to lecture—I remember a lecture by the psychiatrist Jack Barchas, where he discussed the biological basis of schizophrenia and depression. I was blown away that you could explain such complex diseases by imbalances in chemicals in the brain. Scientists and clinicians like these really inspired me.
2. What did you work on during graduate school for your PhD work?
As an undergraduate at Stanford I worked with Roland Ciaranello, who was a psychiatrist and scientist. He didn’t have a PhD but encouraged me to pursue one—[Roland] felt it would have helped him in retrospect. When I got to Harvard Medical School, I started working on dopamine receptor signaling in the gut. At that point I got the feeling that if I didn’t do a PhD I would just be “winging it” with respect to basic science—and that the more rigorous training would be a good idea.
I was intrigued by developmental neurobiology, in particular the work being led by Paul Patterson, Story Landis, Ed Furshpan, and David Potter. I initially worked for Story Landis but then switched to work with Bill Matthew who had trained with Patterson as a post-doc. I studied axonal regeneration. Back then, we were identifying nerve growth promoting factors via monoclonal antibodies. Specifically, I researched how extracellular matrix factors promoted peripheral nerve regeneration. I was asking the question: why can peripheral nerves regenerate pretty well whereas nerve fibers in the CNS do not? So even my PhD was geared towards the clinic, and I had this early desire to help advance new therapies.
3. What attracted you to neurology for residency?
I was relatively open minded but given my interests in neuroscience I knew it had to be psychiatry, neurology or neurosurgery. To be honest, prior to my rotation I wasn’t even sure what a neurologist did—I had a better idea about psychiatry given my undergrad mentor at Stanford. I found that I did not enjoy the practice of psychiatry as much, despite finding the content fascinating. Neurology was amazing: Walter Koroshetz [now NINDS head] was my attending, and he took me under his wing. I also met the well-known neurologist Allan Ropper. I learned from great teachers such as Raymond Adams and C. Miller Fisher. I was mentored by people like Steve Hauser and Bob Brown…and I saw the most interesting patients, and really felt that I was a medical “detective.” Back then, we didn’t have MRIs…we really had to rely on the history and neurologic examination to figure out what was going on.
Another striking thing about neurology: I felt that the level of suffering for these patients was so immense. When the brain is diseased it often prevents the individual from adequately seeking help, caring for oneself or even bonding/communicating with family members. It struck me that it is a double tragedy: being sick and then having the organ responsible for processing and dealing with things [the brain] be dysfunctional. In many ways, I had the most empathy for neurology and psychiatric patients. The biggest drawback [of choosing neurology] was that we had relatively few treatments. However, even in the 1990s we did have some tools: L-DOPA for Parkinson’s, acetylcholinesterase inhibitors for myasthenia gravis, IVIG for Guillain Barre Syndrome [GBS], among other options.
There are also a lot of specialties that have no idea about the brain. In the old days we would spend a lot of time in the ER and admit patients directly from the street. Often, we would be the first physicians to see them after triage. This doesn’t seem to happen as much anymore, but it was certainly a terrific learning experience. Though I hated the hours in the ER, I loved working up totally new patients from scratch. I really felt like a detective at times. In my opinion, this is more fun that just being a consultant to the primary team.
[What were some advances in neurology taking place during your training?]
Towards the tail end of my residency [1990s], the beta-interferon and MS story started to crystallize. A good friend, and fellow MD-PhD [Tim Vartanian], told me about how patients were responding to interferon treatments and that it appeared to be disease modifying. Within MS, we also saw the advent of MRI biomarkers to assess disease progression. I remember seeing a patient at MGH with Steve Hauser, where we were trying to figure out if a patient had a new MS lesion or an exacerbation of a previously existing lesion. When we did an MRI, we saw a new gadolinium enhancing lesion that could explain the patient’s symptoms. This prompted us to treat with high dose steroids, which we may not have done if it was a previously existing lesion.
The effect of beta-interferon on reducing Gadolinium-enhanced T1 lesions as well as new or enlarging T2 lesions is really what made us [neurologists] believe in its effect. Here was objective surrogate [imaging] evidence that correlated with the clinical picture. After seeing this story play out, it really strengthened my interest in going to a place like Biogen.
4. Shortly after becoming an attending neurologist at MGH, you joined Biogen as a Medical Director. What motivated this move?
I was not the brave one: some friends from residency, including Nancy Simonian, made the move first. Nancy went to Biogen two years before I joined. In 1996, I had just become an assistant professor at Harvard with NIH funding (the R29, the so-called FIRST award) for 5 years (along with some smaller grants). Around this time, I saw an ad in the New England Journal for a medical director position at Biogen. The ad specifically called for a neurologist with research experience. I called Nancy Simonian to get more information: she, and others, made it clear that people in industry were incredibly bright and really cared about making drugs for patients—coming from academia I had sometimes heard otherwise. I ended up applying for the medical director job, and Irving Fox was the one who recruited me.
[The transition to industry] proved to be a gradual and somewhat heart wrenching process. I was offered the job at Biogen and declined it at first. During the recruitment process they even sat me at the table with the current CEO during the company holiday party. However shortly afterwards, I had a change of heart and called them to decline the offer. Understandably, they were a bit upset. About a month later [in January], I was at home on the weekend working on a grant. My son came up to me and asked if I wanted to play. I told him I could not because the grant was due soon. He said: “Daddy I don’t want to be a doctor because I’d have to write grants.” I realized that he was really saying: “you are choosing to work rather than be with me.” I felt that just because I wanted to do this “crazy” academic neurology thing, my kid should not have to pay the price. In talking to Nancy [Simonian] about industry, she mentioned that she did not have to work every weekend [if she didn’t want to]. I called Biogen and asked for the job back. They asked: “are you always this indecisive?” Nancy really went to bat for me, and stressed it was a tough decision for me to leave academia. They gave me the job and I started in February of 1998.
5. What were some early lessons learned at Biogen about drug development?
In a company what you work on is not strictly your decision and depends on company strategy and long-term vision. Until you get to be very senior, it is hard to change the direction of the company. I chose Biogen because they had already entered MS and neuroscience with the development of beta-interferon. I trained as a neuromuscular specialist, but always kept an interest in neuroimmunology and MS.
The other major difference is the level of teamwork. In academia you are very focused on your individual output, whereas in industry it is much more about the team. It takes so much expertise to build a drug that teamwork is a necessity—it cannot be “all about you.”
[What was the focus of Biogen when you joined in the 1990s?]
When I joined, Biogen was regarded as an immunology company. They were debating moving into the cardiovascular space. The pipeline featured an anti-CD40 ligand mAb for autoimmune disease, an LFA3 fusion protein for psoriasis and an adenosine A1 receptor antagonist for heart failure. Ironically, there was nothing dedicated to neurology when I started. Richard Flavell had been the head of R&D, so he really shaped the adaptive and innate immunology focus of the pipeline.
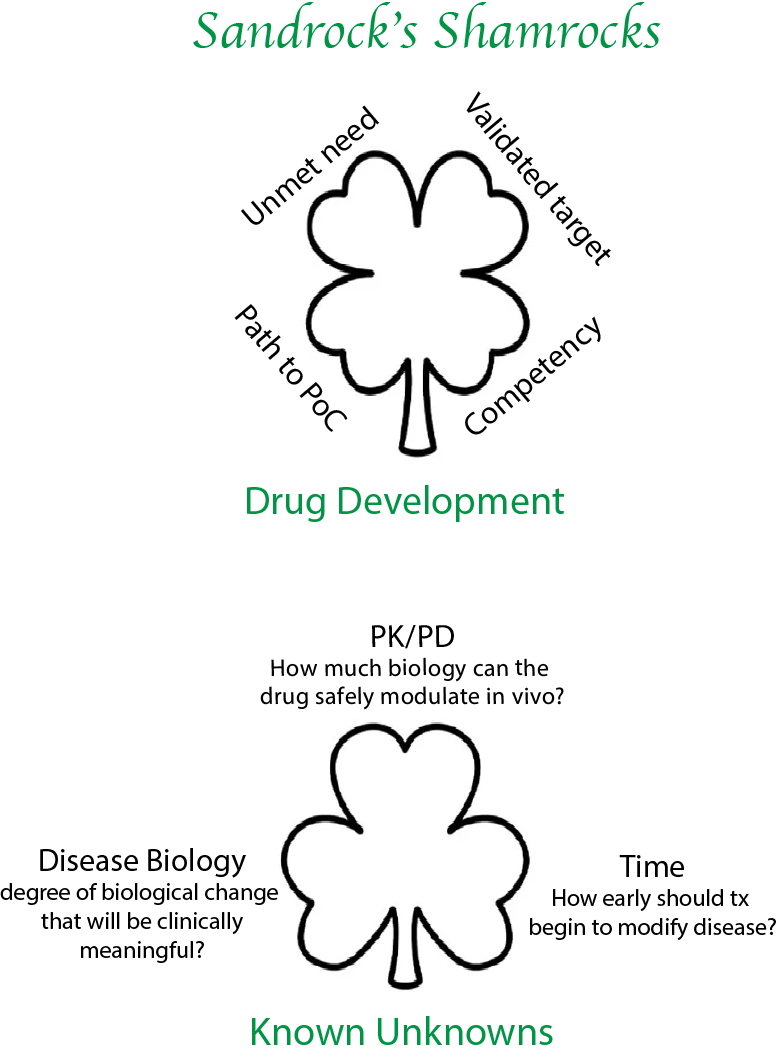
6. You became famous for developing medicines such as Tysabri, Tecfidera and Spinraza. Using these medicines as case studies, what are some checklist items for pushing forward drug development programs?
Sarah Sheikh [now at Takeda] once dubbed my checklist the “Sandrock Shamrock.” When talking about drug development, the first principle is unmet need. This is for several reasons: first, I have to be able to get my team out of bed to work on an important problem. Motivation is a huge factor in drug development.
Further, in diseases of high unmet need, there is often a more flexible regulatory path along with easier patient enrollment. If you are the 15th drug addressing an indication, it becomes harder on all of these fronts. Ultimately (post-approval) there is also less commercial risk if the unmet need is high. The second principle is to find a validated drug target. When we were developing drugs for MS, there was a lot of focus on preclinical animal models—the EAE mouse model for example. Back then (in the 1990s) we didn’t have easy access to human genetics, but now I prioritize human genetic data over animal models. The goal is to find causal biological targets, which is a lot more reliable with human genetic evidence. The third element is path to proof of concept. When you are the head of R&D at a company you do two things: choose [targets/programs] and execute. Even with the right target, if the clinical path to proof of concept is too difficult, the program will languish and the opportunity cost is very high. In neuroscience especially, this can be challenging and is often a huge source of wasted capital for companies. For example, there were so many stroke trials that failed in phase 3 with thousands of patients enrolled. When I first got the job at Biogen, I was lucky to work with Larry Jacobs and Henry McFarland at NIH on MS drug development. NIH was able to do monthly MRIs on patients, and we were able to track how interferon greatly reduced the number of gadolinium enhancing lesions. We also saw that if the patients stopped interferon treatments, these lesions came back. We realized that this imaging biomarker would allow us to run small PoC trials in MS with approximately 50 patients per arm, which could read out in 6 months. We had a path to quickly determining if an agent was promising enough to advance to phase 3 trials.
The fourth element is competency. Do you have the expertise in your company to be able to choose and execute on programs? If you aren’t doing the work in a particular therapeutic area, everything looks good on the surface. And you are prone to make mistakes? The key question is: do you have the competency to evaluate the data and assess if it is interesting, i.e., to choose correctly? Second, do you have the clinical development expertise to execute to de-risk drug candidates efficiently, and ultimately to get the drug approved?
[On other drug development principles]
There are also 3 known unknowns when developing any program, answers to which will be crucial if the program is going to be successful. The first relates to the relationship between drug and biology, i.e., the relationship between pharmacokinetics (PK) and pharmacodynamics [PD]. This is where you start to get an idea of the therapeutic window. How much biology can the drug safely produce in vivo? This is determined for each drug first in animals and then in early human studies. If you are unable to answer this question in animal pharmacology and in early clinical trials, you have not significantly de-risked the program.
The second unknown relates to the relationship between biology and disease. Even though you have chosen to perturb a biological process that is validated by human genetics to be in the causal path to disease, you don’t know how much biological change is necessary to achieve a clinically meaningful treatment effect? For example, how much do we need to lower mutant SOD1 protein to alter disease progression in ALS? This can be learned from other programs with other drugs—this may be where it is an advantage to be a fast follower rather the first in the clinic. The relationship between biology and disease is frequently not linear; there is often a threshold effect, below which biological changes do not produce a discernable clinically relevant treatment effect.
The last known unknown relates to how early treatment should begin. The search for genes that affect the risk of disease may identify biological pathways that are responsible for disease initiation. After disease initiation, other pathways that propagate disease may become more important. In neurology, symptoms often manifest long after disease initiation by which time many cells may have been irreversibly lost. Thus, by the time the patients are diagnosed, disease initiation biology may have given way to other biologic mechanisms that underlie disease progression. For example, by the time patients start anti-amyloid treatment they have had AD pathology for 15 - 20 years or so—I believe this limits the efficacy of such a drug. The same could apply to SOD1 ALS. If we can start treatment in patients before clinical manifestation of disease, we may see larger treatment effect sizes. Early treatment will require novel clinical trial designs and diagnostic biomarkers.
7. What do you see as the biggest barriers to successful neuroscience drug development today?
At the macro level, the biggest current threat to our industry is the concern over drug pricing and reimbursement. In high-risk therapeutic areas like neurology, [R&D and clinical development] is incredibly costly. For an investor, they must see a good return on these high-risk assets, otherwise they will invest elsewhere. In the US, drug prices drive only 11% of health expenditure, but there is disproportionate political focus on regulating prices—in the form of legislation like the IRA. A great primer on this topic is Peter Kolchinsky’s Biotech Social Contract. He makes the point that once drugs go generic, they go generic forever—it is a way to fulfill the social contract after a period of pricing flexibility where the cost and risk of drug development are paid back to investors who took the risk in the first place.
In terms of science, delivery across the BBB has been the biggest obstacle for neurology drug development. ASOs and siRNAs must be injected intrathecally with steep concentration gradients, and relatively poor distribution to deep brain structures. Intraparenchymal injection into the brain also has a ton of safety issues, and similarly poor CNS-wide distribution. I joined Voyager to help make gene therapies more accessible to neurology patients: we are developing key genetic tools for treating neurologic disease.
8. What led you to join Voyager? What about the company mission or internal data was so appealing to you?
While the preclinical data looked promising, I ultimately took a leap in joining Voyager and betting on the company’s capsid technology. Faith and optimism are always part of the equation in biotech…and in many areas of life.
The idea of BBB-penetrant capsids came from the scientist Ben Deverman, originally at Caltech—he named the first of these capsids “PHP,” the initials of his research mentor Paul H. Patterson. Paul was also the scientist that pushed me to pursue an MD-PhD at Harvard and was a cherished mentor for us both. Unfortunately, when Ben did this work Paul was already sick with glioblastoma. Ben told me recently that, tragically, Paul may not have been able to grasp the implications of the novel capsids due to his illness. Ben and I have talked a lot about Paul, because he was both of our inspirations. I told Ben that I know that Paul would have been very proud of his breakthrough research.
But the fact is that the early Ben Deverman capsids worked beautifully in mice, but they only worked in mice and only in certain strains of mice. We now know that the receptor these initial capsids utilized to cross the BBB [Ly6A] was expressed only in rodents. When I saw the Voyager data in non-human primates, I felt that it could be a game changer for patients. Now it could have well been the case that these capsids were using a cynomolgous monkey-specific receptor not found in humans. However, soon after I joined we started seeing cross-species brain penetrance [indicating that the putative receptor was conserved through evolution]. And the Voyager scientists were in hot pursuit of the receptors that mediate BBB crossing. The first such receptor was called Receptor X.
[On finding the “Receptor X” utilized by Voyager capsids]
When I joined the company, I felt that we may not be able to identify receptor X. At Biogen, we ha experience trying, and failing, to identify the receptor that the JC virus (which replicated in the kidney) highjacked to get into the brain to cause PML. However, our scientists [at Voyager] are so good—they were not deterred by my pessimism and ended up finding the receptor. This has been very enabling for the company.
[What does identifying ALPL as one of the capsid receptors do for Voyager?]
Finding that one of our capsid families utilizes ALPL allows us to perform more targeted optimization to develop second and third generation ALPL binding capsids. We can also potentially develop other modalities—antibodies or peptides—that engage this receptor to shuttle cargo (e.g. mAbs or oligonucleotides) into the brain more effectively.
9. Can you discuss some exciting developments in the wholly owned programs? What makes you excited about targeting Tau and AB in AD?
[On Voyager’s anti-amyloid asset in early preclinical development, and Tau mAb]
There are now 3 FDA-approved anti amyloid drugs given intravenously [IV], and hopefully subcutaneous versions of these agents will be coming soon. To develop another IV administered anti-amyloid therapy does not make sense commercially, so this was not our approach. However, under current treatment regimes, AD patients must go every other week or every four weeks for antibody infusions. This can be a huge burden for cognitively impaired individuals. On a personal note, I had to receive IV rituximab for treatment of my non-Hodgkin’s lymphoma. I observed that the IV infusion suites were always packed. The idea of expanding IV mAb infusions to AD patients represents a huge logistical challenge to the healthcare system. Our solution is to vectorize the antibody—use our AAV to enable durable gene expression of an anti-amyloid therapy in the CNS. This would lower the burden on infusion centers and on cognitively impaired patients: We also are working to make a regulatable gene expression platform that could be utilized with our vectorized antibodies—if the patient starts to experience unfavorable symptoms, we want to be able to modulate gene expression to address those symptoms. This is a way to develop a more feasible therapy for patients, while also working on totally new technology.
As we now know, anti-amyloid treatments are not a cure, at least not when used after patients are diagnosed with mild cognitive impairment or early dementia. As such, many of us in the field believe the next best hope is in tau-targeting treatments. Our anti-tau antibody, targeting a C-terminal epitope, is the furthest ahead in development among Voyager’s product candidates. Whereas N-terminal anti-tau antibodies failed to show efficacy in the clinic, there are several companies pursuing mid-domain, MTBR, and C-terminal epitopes. We chose our C-terminal directed antibody because it robustly blocked the spread of human pathological tau in an animal model in which the N-terminal antibodies fail to block the spread. Our goal is to obtain human proof of biology with an IV anti-Tau mAb using tau PET imaging. If that is successful, we then have the option of moving the antibody further in development, vectorizing the antibody with AAV gene therapy, or both. We also have another approach targeting tau in late-stage research utilizing an AAV gene therapy approach whereby we vectorize an siRNA that decreases the expression of tau. We expect this program to enter the clinic in 2026.
We also have a program directed against ALS caused by gain-of-function mutations in SOD1. We anticipate that this will be our first wholly owned gene therapy entering the clinic. We are also advancing two other Neurocrine partnered gene therapy programs. All three programs will employ a novel, BBB-penetrant AAV capsid discovered at Voyager, and we expect INDs in 2025. There is an efficient path in SOD1-ALS to proof of biology (CSF SOD1 levels). as well as proof of concept (plasma neurofilament light). Last year, FDA granted toferseron, an SOD1 lowering anti-sense oligonucleotide, accelerated approval based on plasma NfL, which was deemed a surrogate marker reasonably likely to predict clinical efficacy. Though SOD1 ALS is rare, it validates our gene therapy platform, so has very high strategic value. In our preclinical data, we find that 80% of spinal motor neurons are transduced by our capsids and we can achieve 80-90% SOD1 reduction. So, for our clinical PoC we plan to measure CSF SOD1 levels for target engagement, along with plasma neurofilament as a surrogate. Tofersen has already shed light on the relationship between CSF SOD1 reduction and plasma nFl.
10. What enables IV antibodies to be effective in clearing plaque and target engagement in the CNS, given low CNS distribution?
It's an interesting point. Generally, monoclonal antibodies are very poor are getting across the blood brain brain: the brain to plasma ratio is typically in the 0.1 to 0.5% range. For this reason, one has to give very high doses of peripheral [IV] antibody. The reason why these mAbs can effectively clear plaque is due to the fact that they are incredibly potent. Often these mAbs have picomolar binding affinity. Moreover, they are typically very specific for the intended target. Thus, high concentrations in blood allow for a tiny fraction to gain entry into the CNS is enough to enable target engagement and biological effects. Aducanumab was one of the first pieces of convincing evidence in humans, as evidenced by the robust, dose- and time-dependent clearance of amyloid plaques seen with PET imaging.
11. What makes you most excited about Voyager and its mission over the next 5 years? What are the biggest milestones you hope that the company will achieve?
We want to build a multi-modality neurotherapeutics company going after genetically validated targets. We want it to be heavily focused on neurogenetic medicines, but not just gene therapy [also oligonucleotides targeting RNA]. Even in sporadic disease [science] will eventually identify genetic targets by modern [bioinformatic] methods. We are also collecting some of the best operators to aid our mission. We just hired Toby Ferguson as CMO—he is an MD PhD neurologist with a decade of clinical development experience at Biogen. He led the development of tofersen for ALS, as well as multiple other programs. As CFO, we appointed Nate Jorgensen. Nate is a PhD neuroscientist by training, but garnered extensive experience as a buy side and sell side analyst, and former CFO at a publicly traded company. Voyager aims to collect the best talent and become a leader in neurotherapeutics discovery and development.
12. Any advice for a budding physician-scientist interested in getting involved with drug development or biotech?
Drug development is not taught well, if even taught at all. I was just having this conversation with a colleague in the industry. We agreed that students in medicine and science might benefit from more formal training in drug development.
Right now, my advice would be to “just do it.” Learn by completing internships as a student, and then getting into the industry when you are comfortable taking the leap. I waited until I did a post-doctoral research fellowship, completed residency and clinical fellowship training, and beginning my career as an independent academic neurologist. I feel that I use all of my training every day. But I was 40 years old before I took the leap! My path is not for everybody, but I don’t regret what I did. You should move to industry when you are comfortable, but I would say that there is no substitute for actually joining a team that is trying to make a new treatment for patients suffering from bad diseases. The more innovative the approach, the riskier it is. There is a lot to learn and it can be daunting at times. On the wall behind me, I have a printed out copy of Teddy Roosevelt’s Man in the Arena speech. Do not be afraid to get into the arena and fail if need be. The higher the unmet need, the riskier the program, with greater chance for failure.
I have failed at certain points in my career and even been dragged through the mud for it. I’m likely to fail again: this is the nature of our industry. At certain points, you may feel like giving up. However, all it takes is to meet one patient who has benefited from a drug that you helped to develop. I have met children who would not be alive today without Spinraza. I have danced with MS patients at advocacy events, who would be totally disabled if not for the medicines that we pioneered at Biogen. I still feel like a physician, but my “practice” serves a much larger population than when I was a practicing neurologist.