


Deep Dive: Of Amyloid and Alzheimer's
Briefings on topics in the life sciences and medicine

Introduction
The Alzheimer’s disease (AD) community is at a sobering crossroads. The high-profile failure of Biogen’s Aduhelm, subsequent CMS ruling against anti-amyloid therapies, demise of Roche’s crenezumab, and recent allegations of scientific misconduct, have shaken even stalwart proponents of the “amyloid hypothesis.” It appears that a major pillar of our understanding of AD is weakened, if not crumbling. And yet, decades of genetic, biochemical and mechanistic data support the notion that anti-amyloid therapies could be useful in altering AD pathology. To simply toss out this body of work would—to many patient groups, neuroscientists and clinicians—seem foolhardy.
The stage is set for Biogen/Eisai, Lilly and Roche to unveil the definitive phase 3 results for their anti-beta-amyloid (Aβ) monoclonal antibody (mAb) therapies in Q4 of 2022. To label these trials as “high stakes” is an understatement: AD is the most common neurodegenerative condition, affecting ~ 6 million patients in the U.S. alone. No disease modifying therapies exist. Decades of work from patients, families, advocacy groups, clinicians and scientists have been necessary to run the pivotal trials evaluating these anti-Aβ mAbs.
Clinical failure of these drugs (lecanemab, donanemab, gantenerumab) to slow disease progression will force the AD-community into a hard reset: leading scientists, investors, and pharma companies to pursue alternate therapeutic strategies. Success of these drugs would be a boon to the neurodegeneration field—changing standard of care overnight and filling a huge unmet need for patients.
The moment of truth is almost upon us; before these pivotal phase 3 trials readout, it is important to consider anti-amyloid therapies as a case study. Given what we know now about drugs like aducanumab and crenezumab, how do we view the “amyloid hypothesis” on which these therapies were based? What level of optimism is warranted for anti-Aβ mAbs currently in trials, and what are points of differentiation between these therapies? Are there any useful learnings that have implications for clinical development in other neurodegenerative diseases?
In this deep dive, I will briefly review the central concepts underlying the amyloid hypothesis, and the landscape of past and present anti-Aβ drugs. Phase 2 evidence for the most promising of these drugs, donanemab and lecanemab, will be highlighted. I will also cover several recent developments that add color to the amyloid story: i) CMS’ ruling against this class of medications, ii) the unique trial design used to evaluate Roche’s crenezumab in the API-ADAD study, and iii) the allegations of scientific misconduct in the AD field. Though specific in nature, these events underscore broad issues relevant to future drug development efforts in neurology.
Is it finally time to let the “amyloid hypothesis” and associated therapies “die” so that we can pursue alternate therapeutic strategies? Or will this class of medicines finally prove to be a breakthrough for patients—enabling progress not only in AD but all of neurodegeneration? Let’s dig in.
The Amyloid Hypothesis
The association between Alzheimer’s disease (AD) and amyloid protein stems from observations made in the early 1900s. Oskar Fischer and Alois Alzheimer first described odd pathologic findings in the brains of deceased patients with profound memory loss and dementia. These pathologists noted dense cores (“plaques”) of precipitated protein, adjacent to unhealthy neurons with sickly axonal projections. Activated immune cells next to the plaques suggested that astrocytes and microglia may be trying (and failing) to clear these aggregates.
In the mid-1980s, the main protein constituent of these plaques was identified as beta-amyloid (Aβ), a small protein that had a propensity to aggregate into insoluble masses. The amyloid cascade hypothesis of Alzheimer’s posits that deposition of the Aβ in the brain, and the process of plaque formation, is a central event in disease pathology. More recent neuropathological and longitudinal PET-amyloid imaging studies revealed that Aβ deposition occurs in a stepwise manner—starting in the temporobasal and frontomedial areas, then spreading to the remaining neocortex and finally the striatum. Substantial amyloid pathology is already present in the neocortex decades before overt AD symptomatology. Tau neurofibrillary tangles (NFTs), the other pathognomonic aggregate present in AD, appear after Aβ plaques. However, the spread of Tau pathology has higher clinical correlation to symptom progression than does Aβ, leading some to question the role of amyloid in driving disease pathology. The precise interplay between Aβ and Tau is still the subject of much debate, with some suggesting that amyloid provokes and potentiates NFT pathology.
Skeptics of the Aβ hypothesis state that there is a disconnect between significant amyloid plaque burden and clinical cognitive impairment: for example, there are people who show a high degree of amyloid pathology on post-mortem examination, but did not show any signs of AD during life. Thus, anti-amyloid therapies designed to clear plaques may not provide meaningful clinical benefit. As explored below, there is a growing graveyard of ineffective anti-amyloid therapies (Appendix Fig 2). In this context, skeptics argue that anti-amyloid therapies should be abandoned, and resources allocated to exploring therapies for targeting pathogenic species of Tau or modulating other genetic risk factors like TREM2 and APOE.
Proponents of the amyloid hypothesis argue that since Aβ deposition occurs prior to cognitive decline, early clearance of plaques is required to halt downstream cascades of Tau pathology, neuroinflammation and ultimately neuronal cell death. Further, there is an alternative school of thought that proposes that small oligomers of Aβ peptides (rather than large plaques), are the key neurotoxic species. Aβ plaques simply serve as reservoirs for oligomer release. It was from this vantage points that many hoped treatment of pre-symptomatic genetic AD patients with crenezumab, which has affinity for small Aβ monomers/oligomers, would provide clinical benefit. The unique API-ADAD trial design, which was conducted over the course of 10 years, and study results will be discussed in subsequent sections.
Resources :
Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8(6):595-608. Published 2016 Jun 1. doi:10.15252/emmm.201606210
Karran, E., De Strooper, B. The amyloid hypothesis in Alzheimer disease: new insights from new therapeutics. Nat Rev Drug Discov 21, 306–318 (2022). https://doi.org/10.1038/s41573-022-00391-w
Landscape of Anti-Aβ Therapies
In the past decade, 15 potential therapeutics intended to target the role of Aβ in AD have been tested in phase III trials (Appendix Table 1). These drugs employ several mechanisms of action, including inhibition of enzymes involved in Aβ production and removal of Aβ from the brain using antibodies. All of these therapies have clearly failed to demonstrate clinical efficacy, with gantenerumab, lecanemab, donanemab currently undergoing phase 3 testing (Tables 1-2).
The rest of the discussion will center on using monoclonal antibodies (mAbs) to remove Aβ from the brain. However, a quick note on BACE1 inhibitors: amyloid precursor protein (APP) is cleaved into soluble Aβ by two enzymes, gamma-secretase and beta-secretase (BACE1). Thus, inhibition of secretases, like BACE1, is expected to abrogate Aβ production and slow disease progression. This hypothesis appears to be supported by human genetics: a rare human mutation in BACE1 cleavage site of APP [A673T] results in a 40% decrease in Aβ production in vitro, and a five- to seven-fold reduced risk of developing AD (Jonsson et al., 2012; Maloney et al., 2014). In Alzheimer’s patients, BACE1 inhibitors modestly cleared Aβ plaques from the brain but led to no improvement in cognition. The BACE1 experience highlights two issues: i) it is difficult to meaningfully reduce plaque burden in human AD patients ii) the biology of Aβ is complex and poorly understood—plaque reduction (in symptomatic patients) is not correlated with cognitive improvement.
These issues have also plagued the anti-Aβ mAbs as class of medications. In a recent commentary in Science, researcher and writer Derek Lowe summarizes the AD treatment landscape:
“Every single one of these interventions has failed in the clinic. Every last damn one. If you look for the best outcome of all, actual reversal of Alzheimer’s symptoms, you never see it. No one has, and given the level of neuronal damage, it’s quite possible that no one ever will, unfortunately. What about just slowing down the inexorable progress that the disease seems to show in so many patients? No luck there, either. Compared to control patients, none of these therapies have shown meaningful effects on the rate of decline.” – Dr. Derek Lowe (July 2022)
The only FDA-approved treatment for AD is Biogen’s, now infamous, aducanumab (Aduhelm). What led to aducanumab’s approval? A brief summary of the highly scrutinized history: In March 2019, two identical phase 3 RCTs of aducanumab (NCT02484547, NCT02477800) using Clinical Dementia Rating Sum of Boxes (CDR-SB) as a readout were both terminated following futility analyses. Later that year, Biogen announced that post-hoc analysis of the high dose aducanumab subgroups revealed functional cognitive benefits; they subsequently filed an application for regulatory approval by the FDA. Against the recommendation of its own CNS drugs advisory committee, the FDA granted accelerated approval for aducanumab for
AD treatment in June 2021. This decision was not based on demonstrated cognitive benefits, but rather the effects of aducanumab on a surrogate end point: reduction of amyloid plaque in the brain. Following the accelerated approval of aducanumab, two other mAbs that target amyloid, lecanemab and donanemab, have been granted breakthrough therapy designation by the FDA. The readout of the Ph3 Clarity AD (lecanamab) and TRAILBLAZER-ALZ2 (donanemab) will occur in the Fall of 2022 into early 2023. Like aducanumab, both lecanemab/donanemab are IgG1 subtypes, target overlapping epitopes, and bind Aβ fibrils with higher affinity than monomer/oligomers (Appendix Fig 3). As described in below sections, Roche’s crenezumab is somewhat differentiated on these points.
Donanemab (TRAILBLAZER-ALZ) and Lecanemab both lowered amyloid PET scores significantly over the course of ~18-month phase 2 trials. Below is a summary of these Ph 2 results. Donanemab seems to be the more efficacious of the pair in terms of Aβ reduction and possibly cognitive benefit, but with a slightly higher ARIA risk profile (table 1):


Side effects resulting from mAbs targeting Aβ are non-trivial (Salloway et al., JAMA Neurology 2021). ARIA-E/H occur ~40% of patients on treatment, with 75% of these patients being clinically asymptomatic. Most of the patients experiencing side-effects due to aducanumab treatment (~10%) resolved with halting of medication. However, when weighed against questionable clinical benefit, many consider this risk-profile to be unacceptable. There is some evidence, from phase 3 studies with aducanumab and gantenerumab, to suggest that patients with APOE4 risk variants may benefit more from anti-Aβ drugs, but also bear an increased risk for development of ARIA.
Key Consideration #1: The CMS Decision
In April, CMS decided to limit Medicare coverage of Aduhelm only to patients enrolled in clinical trials. To expand coverage, CMS is demanding additional phase 3 efficacy data. Given the current price tag of Aduhelm ($28k/year, down from $56k), this decision effectively limits access for the average patient. Somewhat surprisingly, CMS decided to expand this ruling to cover anti-Aβ therapies as a class of medications. Thus, CMS is requiring patients enrolled in other anti-amyloid therapy trials (e.g. lecanemab and donanemab) to enter into a registry that tracks and evaluates clinical effects on cognition. CMS will not cover any future approved anti-amyloid therapies unless these drugs meaningfully improve cognitive outcomes. As of July 2022, neurologists can still prescribe a patient aducanumab if they meet criteria; however, patients not on a trial will have to pay out of pocket.
Key questions remain—how meaningful will cognitive improvements need to be in order for anti-Aβ mAbs to garner CMS coverage? If donanemab or lecanemab post ~10% improvement in iADRS or CDR scores, will CMS cover these medication? What about a 5% improvement? Who is exactly is maintaining this anti-amyloid registry, and how will the data be analyzed? How CMS views these drugs, along with the price tags, will determine which patients have access. Heading into the upcoming phase 3 readouts, speculation as to how CMS will respond to pressure from drugmakers, physicians and patient advocacy groups will be top of mind.
Resources:
Salloway, Stephen et al. “Amyloid-Related Imaging Abnormalities in 2 Phase 3 Studies Evaluating Aducanumab in Patients With Early Alzheimer Disease.” JAMA neurology vol. 79,1 (2022): 13-21. doi:10.1001/jamaneurol.2021.4161
Key consideration #2: Roche’s Crenezumab
Crenezumab is a humanized mAb that binds to fibrillar, oligomeric and monomeric forms of Aβ. Though this drug has failed in several phase 3 trials, it is worth reviewing its MOA and the unique API-ADAD trial design. Crenezumab is differentiated from prior anti-Aβ therapies (aducanumab, donanemab, gantenerumab, lecanemab) on several points:
Crenezumab has an IgG4 backbone with reduced effector function and provides lower risk ARIA-E due to decreased vascular amyloid binding.
The drug targets the mid-domain epitopes of amyloid (Appendix Fig 3), which are the hydrophobic regions thought to promote Aβ aggregation. This contrasts with aducanumab, donanemab, gantenerumab, lecanemab, which target the N-terminal domain. An older mAb called solanezumab also binds mid-domain epitopes, and previously failed primary endpoints in four phase 3 trials.
Crenezumab binds multiple species of Aβ (monomers, oligomers, and fibrils): based on in vitro and preclinical mouse data, it binds toxic oligomeric Aβ species and promotes dissociation into soluble monomers. Crenezumab is estimated to have ~ 10-fold higher selectivity towards oligomeric Aβ than other therapies.
Prior to the API-ADAD trial readout, crenezumab failed in CREAD 1 and 2 phase 3 trials conducted in patients with mild AD symptoms:
Crenezumab was tested in two identical phase III trials (CREAD 1 and 2) at 60 mg per kg i.v. Q4W in patients with prodromal or mild AD with mini mental state examination (MMSE) score >22 and a CDR of 0.5 or 1.0 (early symptomatic)
The planned study duration was 2 years, with the primary outcome measure being the change in CDR (coginition). Following interim analyses for futility, both CREAD studies were halted.
In a phase II study, crenezumab was administered for 69 weeks at 15 mg per kg i.v. Q4W, and there was no significant difference in florbetapir amyloid PET between the placebo and dosed groups.
From these clinical data, we can conclude that increasing monomeric Aβ by disaggregating oligomeric forms of Aβ, is not an efficacious strategy in the patient populations tested: no cognitive benefit is observed and unlike other mAbs, Aβ plaques are not effectively cleared.

Rationale for the API-ADAD Trial
Due to its high affinity for Aβ oligomers and ability to bind monomer, it was hypothesized that crenzumab could reduce accumulation of Aβ plaques by enabling the clearance of precursor species. Phase 3 CREAD trials showed an increase in Aβ 1-42 in patient CSF, potentially supporting target engagement (i.e. dissociation of oligomeric species). However, in both CREAD phase 3 trials there was a lack of significant effect on Aβ plaque clearance by PET.
The agent’s much-improved safety profile (reduced ARIA risk) makes it a good candidate drug for prevention in pre-symptomatic and genetically susceptible patients.
API-ADAD is testing a longer treatment period (5+ years) in a genetically defined population (autosomal dominant PSEN1 mutations), prior to AD symptom onset. Thus, an earlier and longer treatment regime may provide clinical benefits not seen in the CREAD 1 and 2 trials
API-ADAD Trial Design
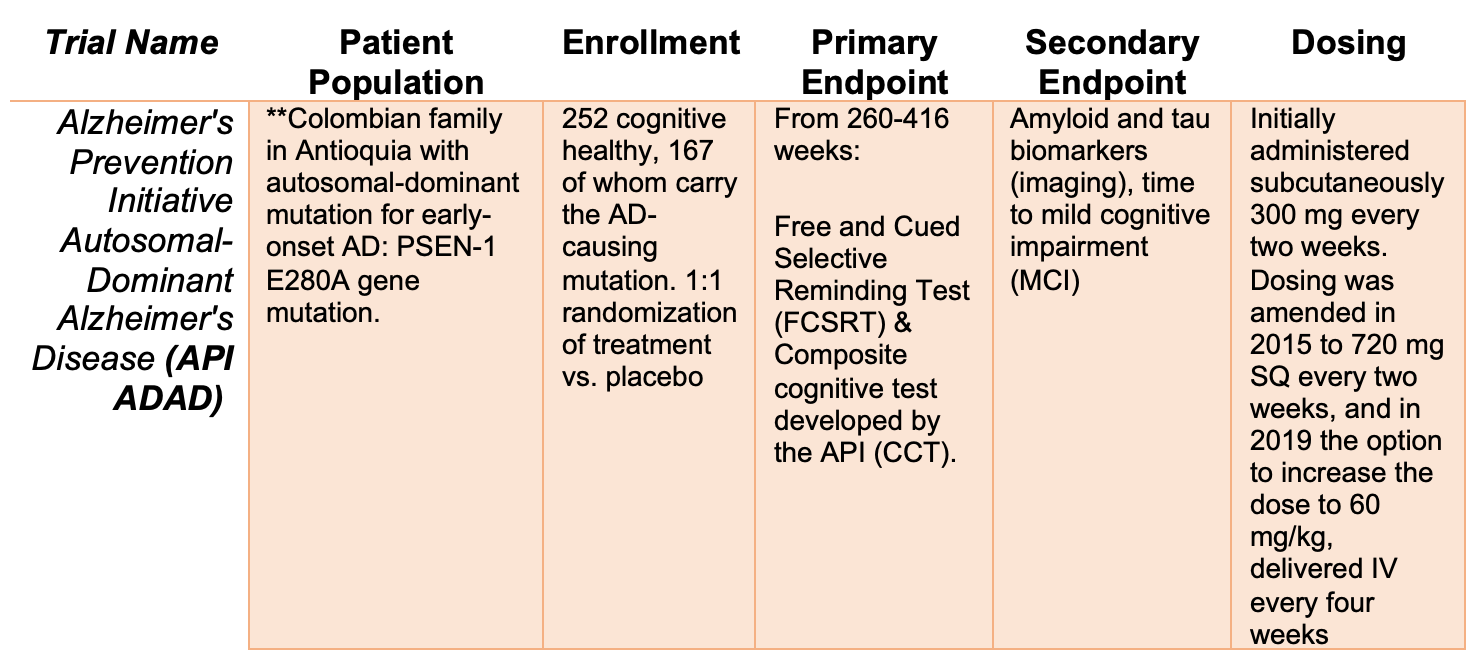
**APOE3: The API-ADAD population is uniquely suited to scientific discovery. Researchers studying this Colombian family found an individual carrying the PSEN1 E280A mutation who was resistant to development of AD. Sequencing of this family member revealed an APOE3 Christchurch mutation (APOE3ch). Recent, post-mortem analysis of this woman’s brain showed high amyloid burden, but decreased hippocampal and cortical Tau pathology. Microglial neuroinflammation was also attenuated. These studies suggest that APOE3ch alters lipid handling in the CNS to impact age of genetic AD onset, severity and disease progression. APOE3 may be an attractive therapeutic target, especially in patients at risk for hereditary AD.
API-ADAD Phase 2 Results
The trial did not demonstrate a statistically significant clinical benefit in either of its co-primary endpoints assessing the rate of change in cognitive abilities or episodic memory function after ~ 5yr treatment with crenezumab.
No additional safety issues or concerns were noted
Full data presented in August 2022 at the Alzheimer's Association International Conference (AAIC). When this data is released by Roche/ACIU it will be helpful to look for the following points:
Target engagement as measured by Aβ 1-42 in patient CSF. If target engagement is demonstrated, it suggests that mid-domain targeting of Aβ is not an effective strategy for reducing plaque burden or improving patient cognition.
Amyloid and Tau PET results. CREAD 1 and 2 showed no reduction in these imaging parameters. However, does longer-term treatment in a PSEN mutant population affect plaque formation? If plaques are cleared and no cognitive benefit is seen this may have implications for other anti-amyloid therapies
Any unexpected side effects or safety issues that could potentially readout onto other programs?
Any (non-sig) trends in cognitive function, as measured by FSCRT or CCT assays?
Resources:
News release (global news)
Key Consideration #3: Allegations of Scientific Misconduct
Last week, Science published an investigation exposing doctored images in a series of influential papers supporting the amyloid hypothesis in AD. This report has attracted a lot of attention, with many claiming it to be the final “nail in the coffin” for Aβ as a therapeutic target.
As discussed above, there are several reasons to doubt the strength of the amyloid hypothesis: dismal clinical data from anti-amyloid therapies and independent evidence supporting other drivers of disease, chief among them. However, the reaction to the recent misconduct is largely overblown – though scientific misconduct is a serious issue, the final accounting on the amyloid hypothesis should be based on assessment of the broader field, prior clinical trials in patients, and the upcoming phase 3 results for donanemab/lecanemab. Below is a summary of the recent scandal, main takeaways and lessons learned. [See Derek Lowe’s excellent article for a more detailed discussion of these issues]
The work in question was conducted by Sylvain Lesné in Karen Ashe’s lab at the University of Minnesota. These studies focused on the isolation and characterization of an aggregated high molecular weight amyloid called AB*56. Lesne and Ashe isolated this species from an aged transgenic mouse model and injected it into healthy young rats – this caused young animals to start exhibiting memory defects. In other words, they suggested that AB*56 is sufficient to cause AD-like pathology. This study has been cited thousands of times, and was the basis of several future studies and grant applications from Lesne and Ashe.
What happened: Matthew Schrag, a neuroscientist at Vanderbilt, came across several Lesné papers flagged on PubPeer – a website highlighting instances of potential misconduct. Analyzing these studies, and the original Lesne/Ashe AB*56 paper, Schrag uncovered many instances of image manipulation. In particular, he found altered images that were intended to show AB*56 is present in brain tissue (using the Western blot method). The Science article illustrates some of Schrag’s analysis—the results appear damning. Schrag has filed a long, and detailed whistleblower report with the NIH – these results will be further investigated, and the original articles hopefully amended or retracted in short order.
Why does this matter? The bulk of research supporting the amyloid hypothesis in AD has nothing to do with AB*56 – many other highly cited amyloid studies (including some from Ashe’s lab) have been reproduced without apparent scandal. In addition, none of the failed or current pipeline drugs targeting Aβ were specifically designed to block AB*56. In his recent article, Derek Lowe argues that while no human trials directly resulted from Lesné’s work, enthusiasm for Aβ agents was definitely affected:
“The AB*56 work did not lead directly to any clinical trials on that amyloid species, and the amyloid oligomer hypothesis was going to lead to such trials anyway at some point. But it certainly did raise the excitement and funding levels in the area and gave people more reason to believe that yes, targeting oligomers could really be the way to go. It’s definitely fair to say that the Lesné work caused these trials to happen more quickly and probably in greater number than they would have otherwise.”
So, what are the key lessons? There are two major takeaways from the AB*56 incident. The first relates to scientific integrity. PubPeer flagged Lesné’s work years before the Schrag investigation—what formal mechanisms exist for following up on credible allegations of misconduct? How can we (financially) support qualified scientists and image experts to do this time-consuming work, and who decides which allegations are worthy of further investigation? To what extent should journals run image analysis AI/ML pipelines and “investigative” quality checks prior to publication? How can we hold unpaid reviewers—lab investigators not often trained in image analysis—responsible for detecting fraud? If image tampering is detected in a published article, what are the next steps; how do we differentiate honest mistakes from bona fide deception? A silver lining: these issues are now forced onto center stage in the scientific community. Since the NIH funds most research in the U.S., the public also deserves convincing answers to all of these questions.
A second takeaway relates to the practice of science. Publishing journal articles is the cheese that makes the academic rat race go round. And why not? Conducting novel research is key for science and society to progress. However, we need well-regarded journals in which to publish negative results. Such “negative studies” are crucial to identify promising future avenues. Harvard neuroscientist Denis Selkoe, a co-author on the Lesné and Ashe AB*56 report, later published work that failed to find this Aβ species in mouse or human brains. However, this negative finding was in the context of other work, which led to its publication in Nature Medicine. We need standalone (ideally peer-reviewed) journals dedicated to publishing high impact negative findings. If these forums did exist, we may not have had to wait almost 16 years after publication of the original AB*56 study for a reckoning.
Deep Dive Takeaways and Conclusions:
Countless trials, novel biomarker development, unique patient stratification approaches, devasting unmet need, bank-breaking costs, cutting-edge science, fake science, shocking FDA approvals, austere CMS rulings, investor lawsuits and high-stakes phase 3 trials. The beta-amyloid field contains multitudes. Below is a summary of the key takeaways, lessons learned and things to watch as we head into Fall 2022:
There is a sobering graveyard of therapies designed to modulate Aβ. Some of these therapies (like gamma-secretase inhibitors) actually worsen disease, while the mAbs effectively clear aggregates but have no apparent effect on cognition. Proponents of the amyloid hypothesis argue that earlier treatment and even more effective clearing of Aβ are needed to realize clinical benefit.
The definitive phase 3 trial results using anti-Aβ mAbs will be revealed in late 2022. Of the current agents under investigation, donanemab (Lilly) is considered the most promising, based on Aβ clearance (PET) and putative effects on cognition (iADRS) in phase 2 testing. Biogen is pinning its hopes on lecanemab; failure of this asset in phase 3 testing will force the company into a hard reset.
The CMS ruling against anti-amyloid therapies as a class of medications is noteworthy—though donanemab and lecanemab will likely be FDA-approved based on Aβ clearance, the bar for CMS coverage will be set by cognitive benefit. The magnitude of clinical benefit needed to garner coverage remains to be seen.
Roche’s anti- Aβ drug crenezumab is another example of a failed AD therapy. Crenezumab targets a different domain of amyloid, and the mechanism of action is differentiated from aducanumab, donanemab and lecanemab. Failure of this drug to effectively clear amyloid or provide cognitive benefit in sporadic AD patients presaged its failure in the API-ADAD trial.
The API-ADAD trial enrolled members of a Colombian family carrying autosomal dominant PSEN mutations leading to early-onset AD. Randomized to placebo or crenzumab, these patients were treated prior to symptom onset and evaluated for over 5 years. Though ultimately a clinical failure for crenezumab, the API-ADAD trial design is intriguing. Treatment of a genetically related family, living in a single community minimizes the effect of genetic background and environment—whether this strategy is feasible to replicate with other anti-Aβ drugs or in other neurodegenerative conditions, remains to be seen. Full data from the API-ADAD trial will be release in August 2022. Specifically, I will look at measures of target engagement (Aβ 1-42), safety issues or trends (not significant) in cognition.
Recent controversy surrounding the AB*56 species is sobering, but not an indictment of the entire body of work supporting the amyloid hypothesis. Other studies and clinical data should be the basis on which we evaluate current and future work in this field. Though amyloid likely plays a (large) role in mediating AD pathology, this does not necessarily validate Aβ as a therapeutic target. Phase 3 results from the anti-Aβ mAbs will put this issue to rest, likely by the end of 2022.
The scandal surrounding scientific misconduct highlights the need for better tools and formal processes to evaluate image tampering. Publication of high-quality “negative results” could also help science progress.
None of the above points are surprising. In the face of current and future negative data, amyloid advocates will argue that earlier treatments are required for clinical benefit. Perhaps this is the case, and Ph3 SKYLINE, AHEAD and TRAILBALZER-ALZ3 trials will shed some light on this matter. However, we should learn from the crenezumab experience; Ph 3 failures in CREAD 1 and 2 predicted negative data in the preclinical genetic populations in Ph2 API-ADAD. Given that the planned Ph 3 trials evaluating donanemab and lecanemab in preclinical patients will be long and costly, perhaps it is better to divert resources to clinical development of other drugs.
Unique genetic cohorts, such as that employed in the API-ADAD trial, may be better leveraged to examine the efficacy of other therapies with unique mechanisms of action. More broadly, it may be time to devote resources away from anti-amyloid into discovering and trialing more effective therapeutic approaches for AD treatment. In the wake of anti-amyloid failures, investors, physicians, and patients will likely be clamoring for new “precision” approaches to targeting AD/neurodegeneration—this could have positive implications for strategies targeting APOE or TREM2 agonism (being pursued by Vigil, Alector and Denali).
Appendix

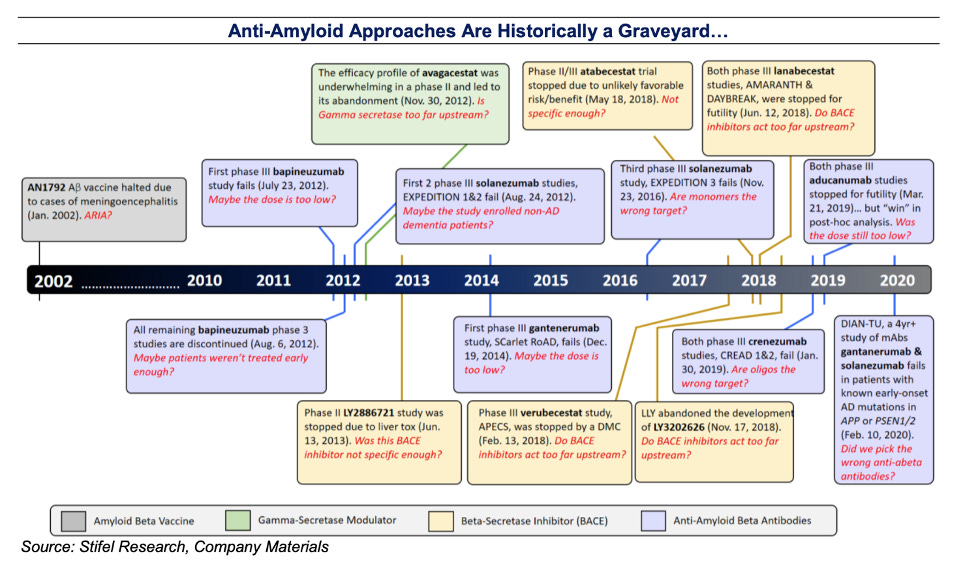


This is a great summary and layout of the timeline. Pubpeer is good for the community (the browser add-on is flagging the references in your write up haha), but it's not addressing the root problem. In my opinion, more tools to evaluate image tampering are great to police, but the problem is in the evaluation of said images. Robust quantitative approaches are needed to substitute traditional methods (i.e. westerns) that lack quantitation. And if images need to be taken, then quantitative approaches should be used to assess the images (numbers don't lie, but I am bias).
Anyways, thanks for a great friday read. keep'em coming!
- Abe