
Deep Dive: Cryptic Splicing and Neurodegeneration
Briefings on topics in the life sciences

The problem: Seminal work by John Q. Trojanowski and Virginia Lee, first described aggregates of a protein called TDP-43 in neurons of patients with amyotrophic lateral sclerosis (ALS) and frontotemporal dementia (FTD). TDP-43 contains domains that can bind DNA and RNA molecules. Normally, this protein resides in the nucleus where it can bind pre-mRNAs and impact RNA-splicing (discussed below). In patients with ALS, the TDP-43 protein is mis localized—it flees the nucleus and forms dense clumps in the cell’s cytoplasm (Figure 1). How mis localization of this RNA-binding protein (RBP) contributes to the fatal neuromuscular disease ALS, is not well understood. A series of recent papers published in Nature, have mapped a role for TDP-43 in modulating “cryptic splicing” of essential neuronal RNAs. Below I will briefly review the molecular biology of RNA-splicing, and the key findings linking TDP-43, cryptic splicing, and neurodegeneration. Promising early-stage companies seeking to modulate splicing in the context of ALS/FTD will be highlighted. Lastly, I’ll offer an opinion on the pros and cons of taking this therapeutic approach in the form of a short investment thesis.

The biology: messenger RNA (mRNA) needs to be processed before translation into protein—one such modification includes the removal or “splicing out” of certain sequences referred to as “introns.” A molecular machine called the “spliceosome” scans RNA and identifies short sequences (splice sites) that delineate which sections to keep (exons) and which to toss out (introns). Yet there is another layer of complexity; short sequences that resemble splice sites occur at many locations within genes. Such sites are not usually recognized by the spliceosome due to repression by a nuclear RNA-binding proteins—molecules like TDP-43 bind close to these sequences and prevent spliceosome access (Figure 2). These hidden or “cryptic” splice sites can become activated in certain circumstances— if for example, if TDP-43 protein is lost from the nucleus. Activation of cryptic splicing can lead to aberrant inclusion of previously “intronic” RNA—such sequences are termed “cryptic exons” (CE). These CEs often contain premature stop codons, leading to production of truncated, non-functional protein. Given that many genes genetically associated with ALS code for RBPs (e.g. TDP-43, SOD1, FUS, ATXN2), precisely mapping dysregulation of this class of proteins impacts RNA splicing and leads to neuronal death, may identify novel therapeutic targets.

Key advances and papers:
These studies sought to understand how loss of nuclear TDP-43 (the most common pathologic hallmark of ALS/FTD) affects RNA splicing in neurons, and how this could lead to disease progression. These works identify dozens of alternatively spliced transcripts in ALS and FTD patients, but focus on two genes that seem impacted the most—STMN2 and UNC13. Loss of nuclear TDP-43 leads to inclusion of cryptic exons in the STMN2 and UNC13A mRNAs. Inclusion of these exons decreases production of mature, functional protein. Crucially, this process appears human-specific and cannot be modeled in mice.
1. Klim et al., 2019 (Kevin Eggan Lab): To model depletion of TDP-43 from the nucleus, Klim et al. used siRNA to knockdown (KD) levels of this molecule in human iPSC cells that that are differentiated into motor neurons (iMNs). By performing RNA-sequencing on these KD cells and controls, they found that TDP-43 depletion leads to a range of alternatively spliced transcripts; among the transcripts with the highest degree of altered splicing (comparing KD and control) was a microtubule-associated gene called STMN2. The team confirmed that alternative splicing of STMN2 transcripts translated to a reduction in protein levels. The group also confirmed that TDP-43 bound directly to STMN2 pre-mRNAs, suggesting a direct role in processing. STMN2 enhances the stability of polymerized microtubules, is enriched in neuronal axons. By restoring levels of functional STMN2, the authors could rescue or prevent the deleterious effects of TDP-43 KD – namely axon growth defects. Further, genetic deletion of STMN2 using CRIPSR-Cas9 editing in iMNs phenocopied the axon growth defects seen in TDP-43 KD neurons. Lastly, the team found that post-mortem spinal cord samples from ALS patients contained cryptically spliced STMN2 transcripts (via RT-qPCR), and that total levels of STMN2 protein were decreased relative to age-matched controls.
2. Brown et al., 2022 (Pietro Fratta and Michael Ward labs): using a CRISPR-based approach to knockdown TDP-43 in cultured iPSC-derived cortical neurons (iNeurons), this group performed RNA-seq on KD and control cells. They validated that STMN2 was alternatively spliced in the TDP-43 KD group (agreeing with Klim et al.), but also detected alternative splicing of another transcript, UNC13A. TDP-43 bound directly to UNC13A transcripts, and its depletion led to inclusion of an UNC13A cryptic exon (CE), and lowered protein levels in cultured cells. UNC13A transcripts containing CEs were found in human FTD and ALS samples, but not in age-matched controls. Perhaps most interestingly, genome wide association studies (GWAS) had previously identified variants in the UNC13A gene (DNA polymorphisms) that increase the chance of developing ALS and FTD. In an elegant minigene assay, Brown et al. then showed that the UNC13A variants associated with disease risk were more susceptible to cryptic exon inclusion following TDP-43 KD. Going one step further, the group found that the UNC13A variants decreased the binding affinity between UNC13A pre-mRNAs and TDP-43. Thus, a model emerges: TDP-43 binds UNC13A in healthy cells. When TDP-43 is depleted from the nucleus, a CE can be included in UNC13A pre-mRNAs leading to decreased protein production (Figure 3). Patients with risk alleles in UNC13A are more likely to have CE inclusion in the setting of TDP-43 depletion – thus perhaps even a slight perturbation of upstream TDP-43 function is enough to induce UNC13A loss.
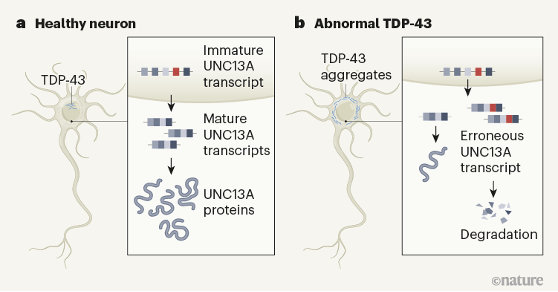
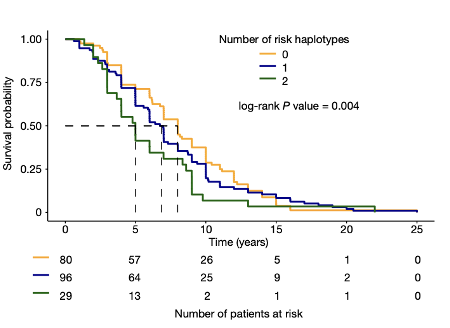
3. Ma et al., 2022 (Aaron Gitler and Leonard Petrucelli labs): Rather than using an in vitro knockdown (KD) of TDP-43 in iPSC-derived neurons, Ma et al., assess the effect of TDP-43 pathology on cryptic splicing directly in humans. In 7 post-mortem FTD or FTD/ALS patient brains, the authors FACS isolated neuronal nuclei that had nuclear TDP-43 (healthy cells),and compared them to neurons with nuclei that did not stain for TDP-43 (suggesting cytoplasmic pathology). Thus, each patient had internal controls; comparing neurons that had lost nuclear TDP-43, with adjacent cells that did not display such pathology (Figure 4). RNA-seq of these sorted neuronal nuclei revealed a host of differentially spliced transcripts between neurons that lacked nuclear TDP-43 and controls, including UNC13A (Figure 4). shRNA-based KD of TDP-43 also led to a loss of UNC13A protein in cultured human cells. As in the Brown et al. study, this group also confirmed that UNC13A transcripts had cryptic exon (CE) inclusion in post-mortem tissue samples from patients with FTD and FTD-ALS. Ma et al. also confirmed that SNPs in UNC13A locus associated by GWAS with development of ALS/FTD promoted CE inclusion in neuronal cells following TDP-43 depletion. Strikingly, a survival analysis found that in FTLD patients, the number of UNC13A risk alleles was negatively correlated with survival; patients with 2 UNC13A risk alleles had a worse prognosis, presumably due to higher rates of CE inclusion and protein depletion (Figure 5).

Remaining questions, not addressed by these studies:
What are the upstream causes of nuclear TDP-43 depletion? The holy grail of neurodegenerative disease research is to find what initiates aberrant protein localization. A very small number of individuals have direct mutations in TDP-43; for the vast majority of patients, the cause of initial cytoplasmic TDP-43 mis localization is opaque. This question was out of the scope of the above studies—nonetheless perhaps there is a role for loss of STMN2 and UNC13A in enhancing or accelerating TDP-43 pathology/misfolding during disease progression.
Though suggestive, these studies do not mechanistically show how loss of STMN2 or UNC13A lead to neuronal demise. Both proteins are hypothesized to be important for neuronal homeostasis at baseline—STMN2 regulates microtubules and possibly maintains axonal structure, while the synaptic protein UNC13A facilitates neurotransmitter release. However, much more about the function of these molecules will need to be uncovered. Prior work suggests that UNC13A overactivity can lead to neuronal death—in this context, inhibiting UNC13A would be beneficial (contrary to the logic of the above papers). The inability to study these processes in mice (which lack these cryptic splice sites) makes it difficult to assess whether depletion of STMN2 or UNC13A alone is sufficient to drive development of ALS/FTD in vivo.
Can STMN2 or UNC13A RNAs containing cryptic exons (CEs) or truncated proteins be used as biomarkers in living patients to measure disease progression (i.e. the extent of TDP-43 pathology)? Are these molecules released from unhealthy neurons, and can they be detected in patient cerebrospinal fluid (CSF) or plasma? Development of such biomarkers will enable target engagement studies for any future therapeutics, stratification of patients for clinical trials, and close monitoring of the natural history of TDP-43 driven pathologies.
TDP-43 pathology is (less commonly) seen in other neurodegenerative conditions like Alzheimer’s and Parkinson’s diseases. To what extent are cryptically spliced STMN2 and UNC13A playing roles in promoting neuronal death in these disease contexts? To what extent does cryptic splicing occur downstream of amyloid-beta, tau or alpha-synuclein driven pathologies?
Competitive landscape: which companies are following up on the findings of these papers to alter TDP-43 driven cryptic splicing in ALS/FTD? How far are these therapies from the clinic?
QurAlis: co-founded by a former member of Kevin Eggan’s group (CEO, Kasper Roet), QurAlis (and partner Q-state biosciences) has two ASO candidates in the pipeline—both designed to reduce alternative splicing of STMN2. As Klim et al., (2019) demonstrated, boosting levels of this protein may provide a survival benefit to motor neurons with TDP-43 pathology. QurAlis is also pursuing an ASO-splice modulating approach in ALS, against an additional “undisclosed” target—one wonders if this target is UNC13A or perhaps another cryptically spliced RNA identified in the more recent Nature papers. QurAlis also has a small molecule program targeting Kv7—a potassium channel that is thought to be pathologically activated in ALS motor neurons.
Maze: co-founded by Aaron Gilter (senior author: Ma et al., 2022), this TRV and ARCH-backed company is likely pursuing an ASO based approach to target cryptic exons in UNC13A pre-mRNA to boost protein levels (although not explicitly stated on company website). Maze is also using an ASO approach to target ATXN2, another RNA-binding protein, which like TDP-43, is genetically linked to development of ALS. Both of these programs are in preclinical development, and will be exciting to watch as leads are identified and announced likely in late ’22 or early ‘23.
Envisagenics: spawned from work at Cold Spring Harbor (Adrian Krainer lab), Envisagenics is using its “SpliceCore” platform to identify alternatively spliced RNAs amenable to intervention in oncology and neurodegeneration. Using RNA-seq data from patients and healthy controls, and a downstream AI/ML pipeline, Envisagenics can identify which alternatively spliced RNAs lead to production of non-functional protein and are thus potential therapeutic targets. Work in neurodegeneration is early (discovery stages), and further along preclinical programs in breast cancer may be validating/de-risking for the SpliceCore approach.
Investment Thesis: will correcting cryptically spliced transcripts in neurodegenerative disease be a promising therapeutic approach in the next 5-10 years? Is the time “right”, or does more basic science need to be done to understand this biology before being leveraged to help patients?
Pros
Human specific biology: since the SOD1 mouse model of ALS was developed in 1994, a dizzying array of potential therapies have shown promise in rodents and failed in the clinic. Researchers, physicians, and investors are skeptical of using these preclinical mouse models to predict success in humans. TPD-43 driven CE inclusion is uniquely human, as these cryptic splice sites are not conserved in mice. Perhaps cryptic splicing events partially explain why conditions like ALS and FTD are so difficult to model in rodents; targeting these human-specific mechanisms of neurodegeneration may ultimately provide the greatest benefit to patients.
Precision approach targeting neurons with pathology: In neurons that are unaffected (i.e. don’t display TDP-43 pathology), cryptic splicing will not occur. In theory, ASO-based therapies designed to bind cryptic splice sites will not impact normal neuronal physiology—instead they will only correct mRNA transcripts in affected cells. This approach differs from a small molecules, which often alter proteins that are present in both diseased and healthy neurons. Note: ASOs can have intrinsic toxicity at high doses, as is hypothesized to underlie the failure of drugs like tominersen in HD.
Enables patient stratification: in the coming years it will be fruitful to overlap alternatively splicing transcriptomic datasets with those from human genetics studies (GWAS). For example, patients with UNC13A polymorphisms are uniquely sensitive to TDP-43 driven cryptic splicing; patients with UNC13A risk alleles may benefit the most from ASO-based therapies designed to correct UNC13A RNAs and restore functional protein levels. Such information will be useful when designing clinical trials.
Cons
Complex biology: in the setting of TDP-43 pathology, STMN2, UNC13A and many other mRNAs are alternatively spliced. Do one or several of these transcripts need to be corrected at once in order to offer therapeutic benefit? If cryptic splicing of many genes mediates pathology, how can we modulate multiple targets with a single therapy? Further, the in vitro preclinical models (iPSC-derived neurons) employed in these studies may not be representative of in vivo biology—that these splicing events do not occur in animal models is a drawback in terms of validating the functional importance of individual RNA splicing targets.
Need for tools to define high value targets: which of the many alternatively spliced RNAs in an ALS/FTD neuron translate to non-functional protein? Is this protein relevant to accelerating neuronal demise? Envisagenics is taking an AI/ML based approach to predict important targets where cryptic splicing leads to loss of functional protein. Though early, these approaches are an important starting point in finding a potential needle in a haystack of alternatively spliced transcripts.
Delivery and efficacy: though ASOs designed to alter splicing have had tremendous success in treating spinal muscular atrophy (SMA; spinraza), hitting other targets in neuro has been challenging. Recent and disappointing results in ALS (SOD1 and C9orf72) and Huntington’s disease (mutant Huntingtin) are likely due to two factors: i) poor ASO penetration and delivery to deep brain structures ii) off-target ASO toxicity. ASOs aimed at modulate alternative splicing in cortical and motor neurons may face similar challenges. How many of these transcripts need to be corrected to supply therapeutic benefit? Can ASOs (with their current chemistries) achieve adequate delivery to cortex and spinal cord to hit this efficacy threshold? Will ASO toxicity (therapeutic index) be a factor? Without adequate preclinical animal models to study these questions, splice modulators in the context of TDP-43-driven diseases may have unpredictable results in the clinic.
Takeaway: Studying how aggregated proteins such as TDP-43 impact cryptic splicing events in ALS, FTD and other neurodegenerative conditions is an area ripe for basic science discovery. The human-specific nature of these alterations (and links to GWAS risk alleles) is intriguing—suggesting that this biology may contribute uniquely to human pathology. However, there are several impediments to early-stage companies leveraging these findings to develop therapeutics. These include: complex biology, lack of preclinical animal models for dose finding and target engagement studies, current lack of biomarkers for clinical trials/patient stratification, and the “traditional” barriers to using ASOs as a therapeutic modality in neurology. Though the next 5 years will likely see many advances made in academia, I think it is unlikely to see any promising “cryptic splicing” therapeutics emerge in this timeframe.
References
Brown AL, Wilkins OG, Keuss MJ, Hill SE, Zanovello M, Lee WC, Bampton A, Lee FCY, Masino L, Qi YA, Bryce-Smith S, Gatt A, Hallegger M, Fagegaltier D, Phatnani H; NYGC ALS Consortium, Newcombe J, Gustavsson EK, Seddighi S, Reyes JF, Coon SL, Ramos D, Schiavo G, Fisher EMC, Raj T, Secrier M, Lashley T, Ule J, Buratti E, Humphrey J, Ward ME, Fratta P. TDP-43 loss and ALS-risk SNPs drive mis-splicing and depletion of UNC13A. Nature. 2022 Mar;603(7899):131-137. doi: 10.1038/s41586-022-04436-3. Epub 2022 Feb 23. PMID: 35197628; PMCID: PMC8891020.
Havens MA, Duelli DM, Hastings ML. Targeting RNA splicing for disease therapy. Wiley Interdiscip Rev RNA. 2013 May-Jun;4(3):247-66. doi: 10.1002/wrna.1158. Epub 2013 Mar 19. PMID: 23512601; PMCID: PMC3631270.
Klim JR, Williams LA, Limone F, Guerra San Juan I, Davis-Dusenbery BN, Mordes DA, Burberry A, Steinbaugh MJ, Gamage KK, Kirchner R, Moccia R, Cassel SH, Chen K, Wainger BJ, Woolf CJ, Eggan K. ALS-implicated protein TDP-43 sustains levels of STMN2, a mediator of motor neuron growth and repair. Nat Neurosci. 2019 Feb;22(2):167-179. doi: 10.1038/s41593-018-0300-4. Epub 2019 Jan 14. PMID: 30643292; PMCID: PMC7153761.
Lipstein N. Mechanism underlying a risk gene in neurodegeneration. Nature. 2022 Mar;603(7899):33-34. doi: 10.1038/d41586-022-00383-1. PMID: 35197585.
Lipstein N, Verhoeven-Duif NM, Michelassi FE, Calloway N, van Hasselt PM, Pienkowska K, van Haaften G, van Haelst MM, van Empelen R, Cuppen I, van Teeseling HC, Evelein AM, Vorstman JA, Thoms S, Jahn O, Duran KJ, Monroe GR, Ryan TA, Taschenberger H, Dittman JS, Rhee JS, Visser G, Jans JJ, Brose N. Synaptic UNC13A protein variant causes increased neurotransmission and dyskinetic movement disorder. J Clin Invest. 2017 Mar 1;127(3):1005-1018. doi: 10.1172/JCI90259. Epub 2017 Feb 13. PMID: 28192369; PMCID: PMC5330740.
Ma XR, Prudencio M, Koike Y, Vatsavayai SC, Kim G, Harbinski F, Briner A, Rodriguez CM, Guo C, Akiyama T, Schmidt HB, Cummings BB, Wyatt DW, Kurylo K, Miller G, Mekhoubad S, Sallee N, Mekonnen G, Ganser L, Rubien JD, Jansen-West K, Cook CN, Pickles S, Oskarsson B, Graff-Radford NR, Boeve BF, Knopman DS, Petersen RC, Dickson DW, Shorter J, Myong S, Green EM, Seeley WW, Petrucelli L, Gitler AD. TDP-43 represses cryptic exon inclusion in the FTD-ALS gene UNC13A. Nature. 2022 Mar;603(7899):124-130. doi: 10.1038/s41586-022-04424-7. Epub 2022 Feb 23. PMID: 35197626; PMCID: PMC8891019.
Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006 Oct 6;314(5796):130-3. doi: 10.1126/science.1134108. PMID: 17023659.